1. 什么是价键理论?
这一讲主要向大家简单介绍价键理论,使大家对价键理论有个基本印象。其中可能会涉及一些大家比较陌生的基本概念,不用紧张,我们会在后续章节中详细阐述。
1.1. 经典价键理论简介
首先,让我们先思考,并试着回答以下问题:
思考
H2 分子是如何成键的?
关于这个问题,价键( VB )理论和分子轨道( MO )理论的解释是不太一样的。主流的分子轨道理论的解释是:
两个 H 原子的 \(1s\) 原子轨道由于能量相近,对称性匹配,因此可以线性组合为两个分子轨道:一个
成键轨道和一个反键轨道。这两个分子轨道都是离域的,且相互正交。成键轨道的能量低于原子轨道能量,反键轨道的能量高于原子轨道能量;
H2 分子的两个电子按照能量由低到高的顺序填充分子轨道,因此两个电子都填充在成键轨道上,而反键轨道没有电子。由此,两个H原子之间形成化学键,体系能量低于两个H原子的能量。
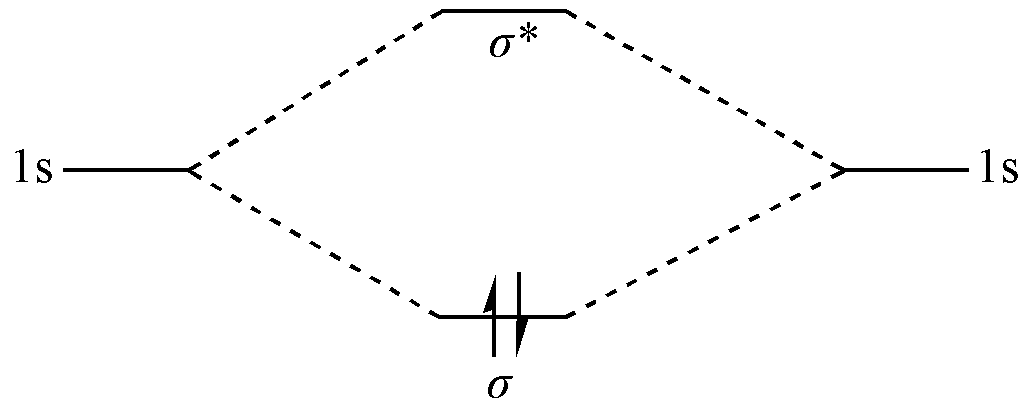
图 1.1.1 分子轨道理论中 H2 的成键机制
可以看到,在分子轨道理论中,电子是天然在体系中离域的。 定域 只是离域的一种特例。化学键的形成是因为成键轨道上的电子多于反键轨道上的电子。
提示
当我们说一个轨道是 定域 或 离域 时,是指当电子填充到这个轨道中后,电子云的范围。当一个轨道是 定域 的,意味着对应的电子云只能出现在某一个区间;如果一个轨道是 离域 的,则对应的电子云可以出现在体系中的任何地方。
在价键理论中,H2 分子的成键机理则是:
两个 H 原子轨道是 定域且非正交 的,每个轨道定域在各自的 H 原子上。电子在体系中以自旋反平行的形式进行配对;
H2 分子的 2 个电子在两个 H 原子之间 共享 。由此,电子在两个 H 原子的定域原子轨道中有多种可能的占据形式。电子可能分别占据两个原子轨道,或者同时占据某个原子轨道。这些不同的占据形式构成了不同的 价键结构 。
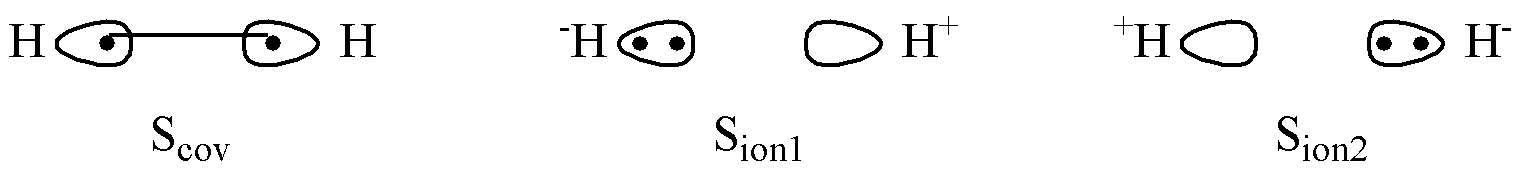
根据量子力学原理,这些价键结构在 H2 分子的最终形态中分别以一定的几率存在,形成 共振 。同时,在电子分别占据两个轨道的情况下,两个自旋反平行的电子还存在着 交换 的现象。
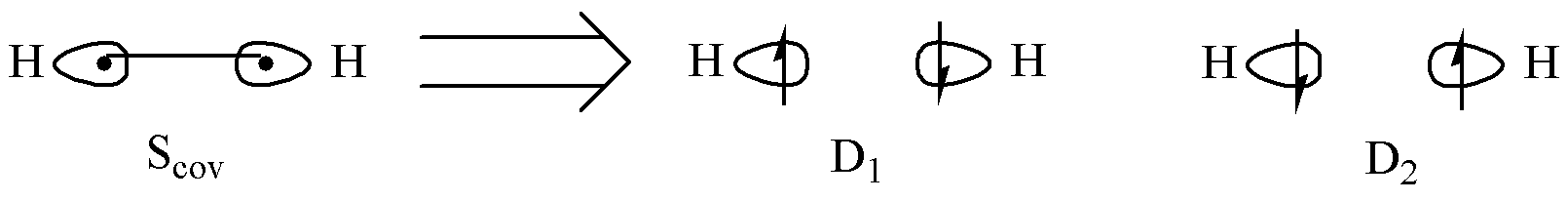
注意
由于价键结构和共振密切相关,因此价键结构有时也被称为共振结构。在接下来的内容中,我们不会刻意区分二者。
可以看到,在价键理论的视角中,化学键是定域的;离域是定域的一种特殊形式。化学键籍由两个自旋反平行的电子因在成键原子之间配对而产生的 交换 和 共振 得以形成。
我们可以将这些差别归纳为下面这个表格:
VB |
MO |
|
|---|---|---|
成键是否定域 |
是 |
否 |
轨道是否正交 |
否 |
是 |
成键来源 |
电子交换与结构共振 |
成键轨道电子多于反键轨道电子 |
到目前为止,对于初学者而言,可能已经开始被那些诸如“结构”、“共振”之类的概念搞糊涂了。没关系,先尝试接受它们,我们将在后面的章节中详细阐述它们的确切含义。
好了,通过这个例子,我们相信,大家已经基本了解了经典价键理论的特点。简单来说,经典价键理论就是一种采用 定域非正交原子轨道 ,通过电子在原子轨道中的不同占据形式产生 共振结构 来描述化学键,并解释化学现象、阐述化学反应机理的一种化学键理论。
1.2. 价键理论的特点:优势与劣势
原子间如何成键一直是化学家们关心的核心问题。自从 Lewis 那篇著名的论文发表之后, 电子配对成键 的概念逐渐深入人心。而 Heitler 和 London 关于 H2 成键的研究工作以及 Pauling 共振论的提出标志着价键理论的最终成型。价键理论的最大优势在于它的直观性。价键理论的 定域成键 概念符合人们对化学键的直观理解,价键结构则是这种成键理念的具象化。通过这种具象化的结构,人们可以非常方便地探讨和理解化学反应中体系电子结构(如原子轨道、电子占据、化学键极性等)的变化。这是分子轨道理论所无法比拟的。因此,早年(上世纪 50 年代之前)价键理论是人们讨论化学反应机理,解释化学现象的主要工具。
然而,随着计算化学的发展,分子轨道理论逐渐取代了价键理论成了人们的主要工具。其中最主要的一个原因就是价键计算的复杂性。即使对于一个 H2 分子,价键理论也需要数个价键结构(我们会在后续章节中详细讨论给定体系的价键结构)来描述化学键。加上价键理论采用的定域非正交轨道使得计算量进一步加大。这导致价键理论的计算量随着化学问题涉及的化学键增加而急剧增加。在早年,如此庞大的计算量常常使得定量计算无法进行。与此同时,分子轨道理论依然保持着简单的形式和可接受的计算量。这使得价键理论逐渐沦为了一种 仅用于定性描述 的工具。期间有一些 改进的 价键理论方法,比如著名的广义价键( GVB ),自旋耦合价键( SCVB )等。和以往的价键方法不同,这些价键方法采用了离域的轨道和一些正交的性质来降低计算量。然而,也正因为轨道的离域,这些方法在降低计算量的同时也降低了价键结构对于化学键描述的精确性。我们通常将这些采用离域轨道的价键方法称为 现代价键理论 ,而将采用定域轨道的价键理论方法称为 经典价键理论 。
好在随着计算机技术的发展,经典价键理论的计算量逐渐变得可以接受。上世纪 80 年代起,随着 价键自洽场( VBSCF ) 方法的发展,经典价键理论的从头算变得可行。之后 呼吸轨道价键( BOVB ) , 价键组态相互作用( VBCI ) , 价键二级微扰( VBPT2 ) 等方法的发展使基于经典价键理论的精确计算成为现实。
1.3. 对价键理论的误解
分子轨道理论取代价键理论成为主流化学键理论的另一个重要原因就是人们对价键理论,特别是共振论的误解。一个非常经典的例子就是对 O2 基态的解释。传统共振论认为,O2 成键的共振结构应该类似如下模式:

图 1.3.1 传统共振论中 O2 的成键机制
在这种模式中,O2 总共有两个键:一个 \(\sigma\) 键和一个 \(\pi\) 键。按照这种成键模式,O2 应该是没有未成对电子的,呈现反磁性。然而,实验表明,O2 分子的成键虽然是双键,但是基态 O2 是顺磁性的,这意味着 O2 分子中存在着未成对电子。于此同时,分子轨道理论也给出了另一种成键模式(我们忽略了对成键分析没有影响的 \(1s\) 和 \(2s\) 轨道部分):
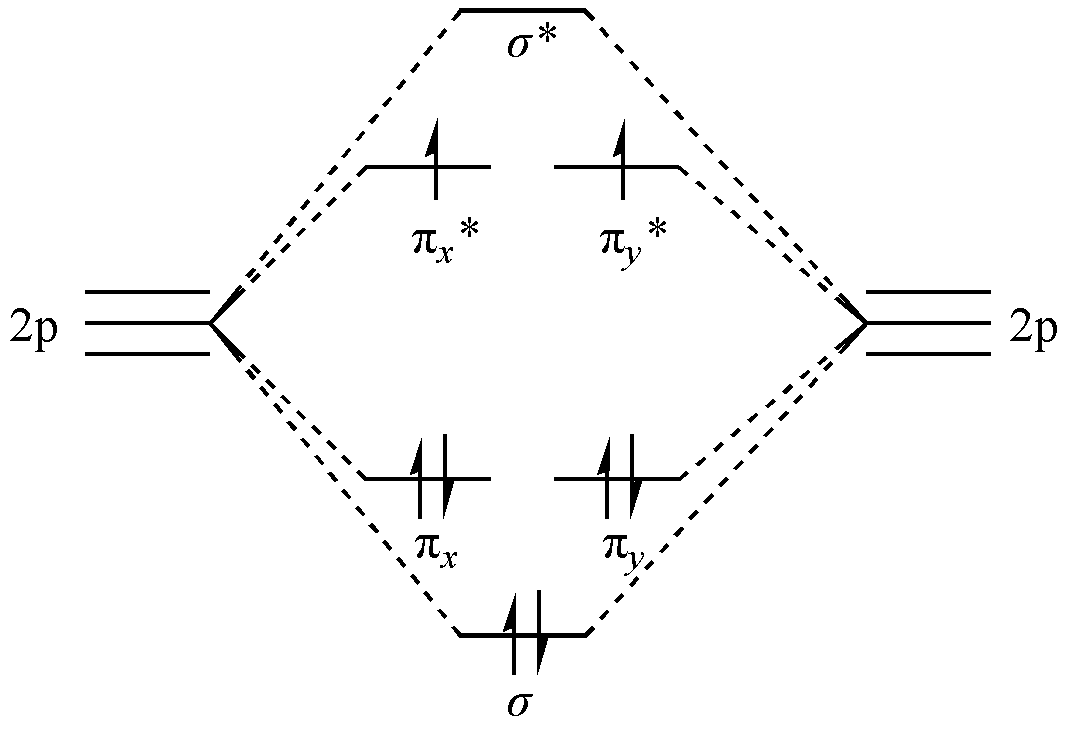
图 1.3.2 分子轨道理论中 O2 的成键机制
如 图 1.3.2 所示,6 个 p 轨道线性组合为分子轨道,8 个 p 电子按照能量最低原理和洪特规则进行填充,正好构成了三重态的基态,总键级为 2 ,表明 O-O 之间的化学键为双键。
分子轨道理论完美解释了O2 分子的成键,这也让化学家们对价键理论的正确性产生了怀疑。造成这种情况的原因是当时人们试图用简单的共振形式来描述 O2 分子的成键导致的。实际上,Pauling 在早年就曾认为,O2 分子 \(\pi\) 方向上的成键不应该是简单的单键,而是两个3电子2中心键,如下图所示:

图 1.3.3 价键理论中 O2 的成键机制
2007 年,厦门大学苏培峰等人 对 O2 分子的价键计算证明了这一点。由此可见,价键理论依然是有效且正确的。
1.4. 什么时候需要用价键理论?
虽然这个课程是关于价键理论的,但是我们依然在这里强调,不是所有计算都需要价键理论。如今,分子轨道理论所展示的图像已被普遍接受,拥有丰富的计算方法、大量的计算软件以及高效的计算效率,对热力学性质、化学反应过程、激发态和光谱等都可以给出令人满意的结果,因此没必要为了标新立异采用价键理论。除非你的研究内容和化学键紧密相关,同时分子轨道理论无法给出令你满意的解释,你才需要考虑价键理论。