6. 激发态的计算与分析
到目前为止,我们关注的都是基态的计算。在实际科研任务中,分子不可能总是处于基态,人们的研究兴趣也不可能一直集中于分子的基态性质。因此,我们有必要在这一讲中简单讲一下如何进行激发态的计算。
6.1. MO 和 VB 中激发态的不同图像
在 MO 理论中,原子轨道经过线性组合得到分子轨道。分子轨道具有各自的能量,电子填充轨道遵循能量最低原理,即按照能量由低到高的顺序依次填充。如果遇到能量简并的情况,那就依据洪特规则,优先以自旋平行的方式填入各个简并轨道,然后再填满。按照这种法则得到的电子态,一般都是基态,因为此时空轨道的能量都比占据轨道高。如果电子填充轨道的时候没有遵循这些规则,比如填充简并轨道的时候没有遵循洪特规则,而是先填满了某个简并轨道,其它兼并轨道依然是未占据状态;或者没有遵循能量最低原理,优先填入了更高能量的轨道,更低能量的轨道反而是未占据或者未占满的状态,都会使得分子的能量升高。这种更高能级的状态就是所谓的 激发态 。这种状态是不稳定的,通常都是基态分子中的电子由于得到了某种形式的能量,比如光照,从而使电子跃迁到了更高能量的轨道或者组合形式而得到的。MO 理论中,分子的激发能可以粗略地用 Koopmann 原理进行估算,即
实际计算中,由于电子激发后,轨道会由于电子占据情况的改变而发生弛豫,实际的激发能还需要考虑轨道弛豫的影响,需要进行自洽场迭代优化。
在 VB 理论中,分子的波函数取决于两个方面:构成共振结构的轨道,以及共振结构间的线性组合。不同的轨道赋予了共振结构不同的物理含义和化学图像,不同的组合系数赋予了分子不同的性质和状态。组合系数通过求解久期方程得到。对于一个有 n 个结构的分子,久期方程对应于一个 n 阶广义本征方程,总共可以得到 n 个根。其中第一个根提供了最低的能量对应的组合形式。当轨道占据遵循能量最低原理时,第一个根提供的就是我们通常需要的基态。如果我们不选取第一个根,或者电子占据的不都是能量最低的轨道,我们得到的结构的组合就会发生变化,我们就得到了激发态。 我们从这里可以看到,VB理论的激发态计算有两个维度:轨道的选取和组合系数的变化。由于轨道的选取关系到共振结构具体的物理含义和化学图像,请参照我们的 节 2 和 节 3 内容。这一讲我们关注的是组合系数的变化对激发态的影响。
6.2. 如何在XMVB中计算激发态
6.2.1. 获取特定激发态
XMVB中提供了选取不同的久期方程的根的关键字 NSTATE=IROOT ,其中 IROOT 是一个非负整数,表示第几个激发态。 NSTATE=1 表示此时程序将选取第二个根来获得第一激发态的计算, NSTATE=2 表示第二激发态的计算,以此类推。 NSTATE=0 表示将进行基态(第零激发态)的计算,也是这个关键字的默认值。
我们先以H2 为例,看看 NSTATE=IROOT 的效果。默认情况下,XMVB 进行的是基态计算。对于H-H键长0.7 Å ,cc-pVDZ 基组的 HAO 计算,我们得到的能量是 -1.14386 a.u. ,3 个价键结构的系数和权重如下:
1 ****** COEFFICIENTS OF STRUCTURES ******
2
3 1 -0.84426638 ****** 1-2
4 2 -0.09228443 ****** 1 1
5 3 -0.09228443 ****** 2 2
6
7
8 ****** WEIGHTS OF STRUCTURES ******
9
10 1 0.84327776 ****** 1-2
11 2 0.07836112 ****** 1 1
12 3 0.07836112 ****** 2 2
可以看到,此时是共价结构占绝对主导的。如果我们加上了 NSTATE=1 ,最后的能量就变成了 -0.38420 a.u. ,相应的结构系数和权重也变成了:
1 ****** COEFFICIENTS OF STRUCTURES ******
2
3 1 -0.00000000 ****** 1-2
4 2 1.51521437 ****** 1 1
5 3 -1.51521436 ****** 2 2
6
7
8 ****** WEIGHTS OF STRUCTURES ******
9
10 1 0.00000000 ****** 1-2
11 2 0.50000000 ****** 1 1
12 3 0.50000000 ****** 2 2
可以看到,此时的波函数中已经没有了共价结构,离子结构的系数也从相同变成了相反。此时得到的就是第1激发态对应的结构系数。
接下来我们来看一个复杂一点的例子:O2 。O2 的三重态计算需要 12 个共振结构,单重态计算需要 10 个结构。三重态的 12 个结构见 图 6.2.1 。其中最重要的是 T1 和 T2 这两个结构。
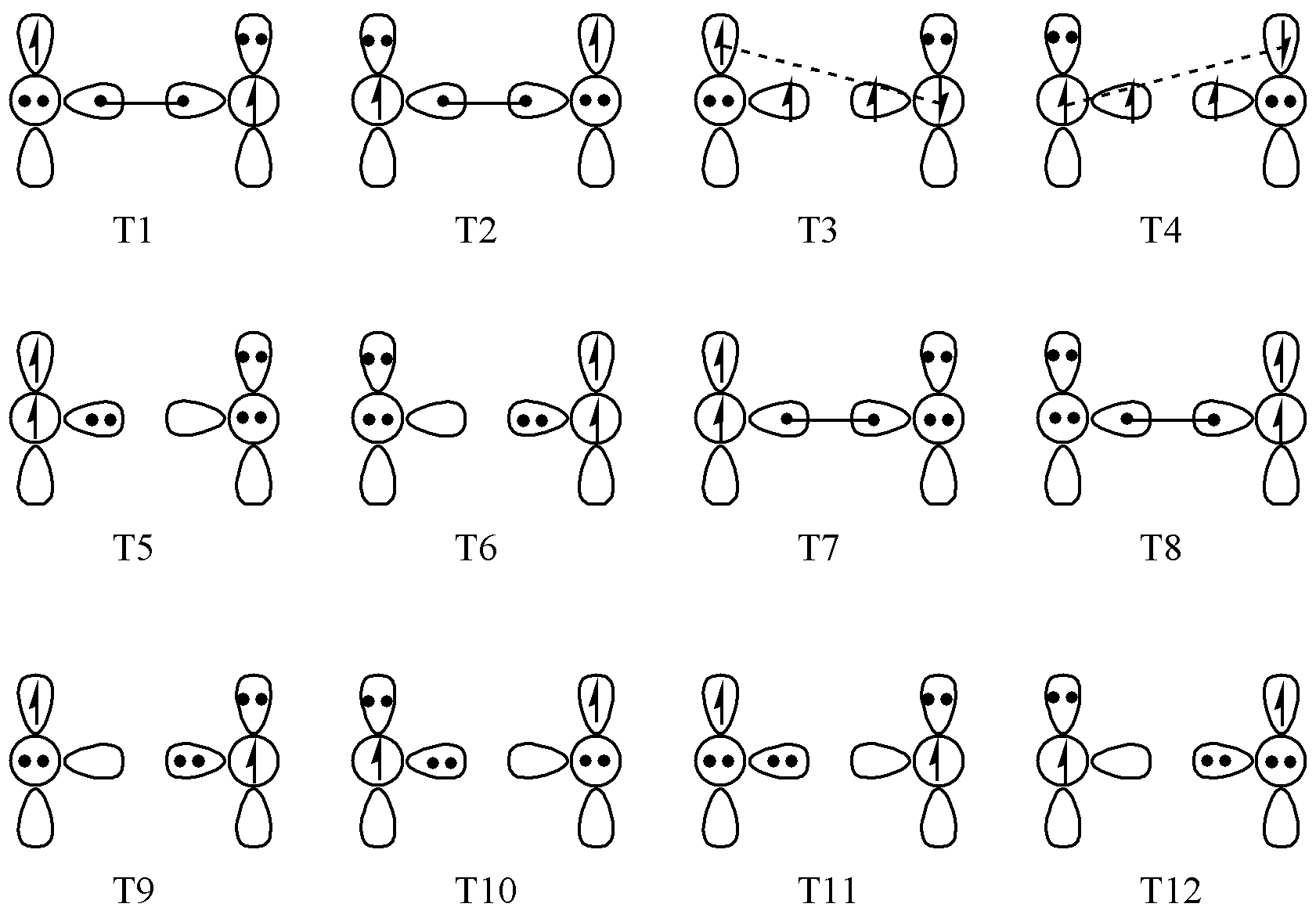
图 6.2.1 O2 三重态的12个价键结构
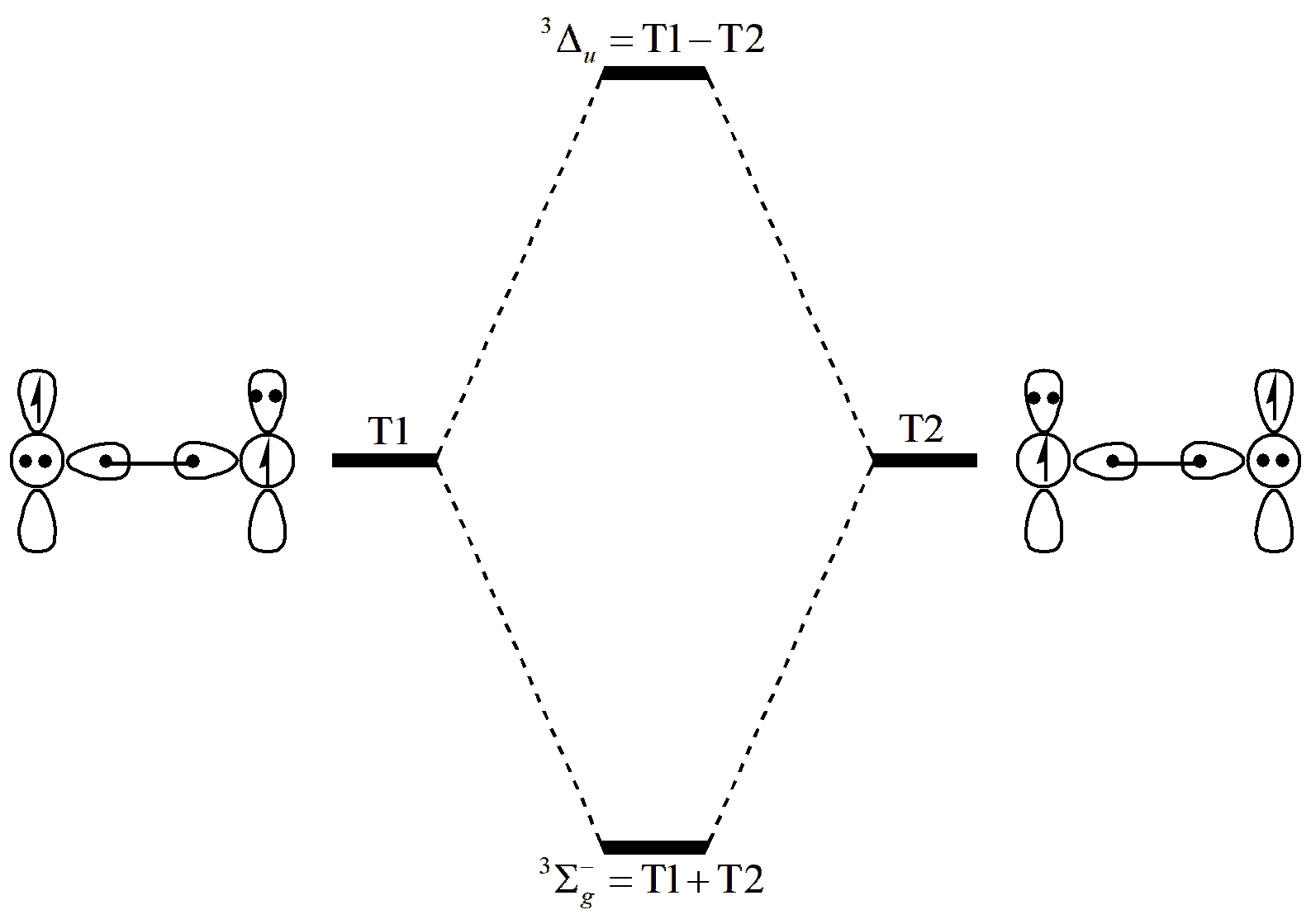
图 6.2.2 O2 三重态的基态和激发态
从 图 6.2.2 可以看到, T1 和 T2 的不同组合方式对应了不同的电子态。对于基态,两个结构的系数是相同的;对于激发态,组合系数是相反的。
为了得到三重态的激发态,我们可以利用 NSTATE=1 来计算三重态的第一激发态,如下:
1O2 L-VBSCF
2$ctrl
3vbscf nstr=12 nao=6 nae=8 iscf=5 iprint=3
4orbtyp=hao frgtyp=sao nmul=3 itmax=200 nstate=1
5int=libcint basis=cc-pvdz
6guess=read
7$end
8$str
91:4 7 7 10 10 5 6 8 9
101:4 8 8 9 9 5 6 7 10
111:4 7 7 10 10 8 9 5 6
121:4 8 8 9 9 7 10 5 6
131:4 7 7 9 9 5 6 8 10
141:4 8 8 10 10 5 6 7 9
151:4 8 8 10 10 5 5 7 9
161:4 7 7 9 9 6 6 8 10
171:4 8 8 9 9 5 5 7 10
181:4 7 7 10 10 6 6 8 9
191:4 7 7 10 10 5 5 8 9
201:4 8 8 9 9 6 6 7 10
21$end
22$frag
231*6
24spzdxxdyydzzfzzzfxxzfyyz 1
25spzdxxdyydzzfzzzfxxzfyyz 2
26pxdxzfxxxfxyyfxzz 1
27pxdxzfxxxfxyyfxzz 2
28pydyzfyyyfxxyfyzz 1
29pydyzfyyyfxxyfyzz 2
30$end
31$orb
321*10
331
342
351
362
371
382
393
404
415
426
43$end
44$geo
45O 0.0 0.0 0.0
46O 0.0 0.0 1.2
47$end
48$gus
49 8 8 8 8 8 8 3 3 3 3
50 1.0019774038 1 0.0065168066 2 0.0001788772 3 0.0010309980 6
51-0.0003633198 9 -0.0027414664 10 -0.0030686413 13 -0.0030026561 15
52 1.0021643892 16 0.0072505362 17 0.0002382456 18 -0.0011188449 21
53 0.0003834680 24 -0.0030102480 25 -0.0031503232 28 -0.0032525782 30
54 0.0982223750 1 -0.2504910231 2 -0.8206750714 3 -0.0401288970 6
55-0.3250457745 9 0.0663093980 10 0.0645475585 13 -0.0255293114 15
56 0.1023941578 16 -0.2356452123 17 -0.8258612653 18 0.0419646005 21
57 0.3491578062 24 0.0683659578 25 0.0684486346 28 -0.0278018600 30
58-0.0534466425 1 -0.2957625003 2 -0.0041914149 3 0.2677956458 6
59 0.7578794938 9 -0.0782023209 10 -0.0786400588 13 0.0255903120 15
60-0.0521455763 16 -0.3057336084 17 -0.0199773005 18 -0.2554552086 21
61-0.7557006120 24 -0.0788914259 25 -0.0791889215 28 0.0231651215 30
62 0.5315910156 4 0.6213802169 7 0.0117253831 12
63 0.5179245322 19 0.6342323659 22 -0.0135520711 27
64 0.5280659286 5 0.6247144659 8 0.0122018946 14
65 0.5244186959 20 0.6281355341 23 -0.0136783062 29
66$end
相对于基态的计算,激发态计算的文件仅仅多了一个 NSTATE 关键字。我们可以将 NSTATE=1 去掉实现基态的计算。最终,我们得到了 表 6.2.1 的计算结果:
结构 |
基态 |
激发态 |
||
|---|---|---|---|---|
系数 |
权重 |
系数 |
权重 |
|
T1 |
-0.343 |
0.220 |
-0.492 |
0.327 |
T2 |
-0.343 |
0.220 |
0.492 |
0.327 |
T3 |
-0.018 |
0.000 |
-0.036 |
0.001 |
T4 |
0.018 |
0.000 |
-0.036 |
0.001 |
T5 |
0.166 |
0.073 |
0.000 |
0.000 |
T6 |
0.166 |
0.073 |
0.000 |
0.000 |
T7 |
0.178 |
0.091 |
0.000 |
0.000 |
T8 |
0.178 |
0.091 |
0.000 |
0.000 |
T9 |
-0.125 |
0.058 |
-0.180 |
0.086 |
T10 |
-0.125 |
0.058 |
0.180 |
0.086 |
T11 |
-0.125 |
0.058 |
-0.180 |
0.086 |
T12 |
-0.125 |
0.058 |
0.180 |
0.086 |
从计算结果中可以看到, T1 和 T2 两个结构的系数在基态和激发态中的系数组合确实发生了变化。同时我们可以看到,无论从系数还是权重来看,激发态相对于基态更集中于 T1 和 T2 ,其它结构在总波函数中的贡献均有下降。O2 三重态的第一激发态更主要由 T1 和 T2 两个结构组成。
6.2.2. 态平均计算
我们在进行激发态计算的时候,常常会使用“态平均”。所谓态平均,就是我们不是优化某一个特定的电子态,而是将数个我们感兴趣的电子态能量按一定的权重进行组合,利用自洽场迭代优化,使得组合后的总能量达到最低:
其中 \(E\) 为总能量, \(E_i\) 为参与态平均的某个态的能量。 \(w_i\) 为事先拟定的权重,在整个迭代优化过程中保持不变。
在XMVB中,我们可以利用 WSTATE 关键字进行态平均计算。WSTATE 关键字的语法为:
WSTAE( start )= \(w_{\textrm{start}}\), \(w_\textrm{start+1}\), ...
其中括号中的 start 表示起始态的编号,基态为1,第一激发态为2,以此类推。等号后以逗号为间隔,可以输入多个对应的权重。例如:
WSTATE(1)=0.4,0.6
表示进行一个两态的态平均计算,其中基态的权重为 0.4 ,第一激发态的权重为 0.6 。WSTATE 关键词也可以多个叠加使用,比如:
WSTATE(1)=0.5,0.5 WSTATE(5)=0.4,0.6
表示态平均计算涉及 4 个态:基态(权重 0.5 ),第一激发态(权重 0.5 ),第四激发态(权重 0.4 )和第五激发态(权重 0.6 )。需要注意的是,此时权重之和已经大于 1 了。XMVB 会自动将总权重进行归一,因此上述关键词等同于:
WSTATE(1)=0.25,0.25 WSTATE(5)=0.2,0.3
用户只需要关心不同态之间权重的比值即可。WSTATE 的默认值为
WSTATE(1)=1.0,0.0,0.0, ...
即只进行基态的优化。态平均计算可用于 VBSCF 和 BOVB 计算。 我们以 H2 为例,进行一个态平均计算。这个算例涉及基态和第一激发态,对应的权重分别为 0.7 和 0.3 。输入文件如下所示:
1H2 State average
2$ctrl
3vbscf
4str=full nao=2 nae=2 iscf=5
5orbtyp=hao frgtyp=atom
6int=libcint basis=cc-pvdz
7wstate(1)=0.7,0.3
8$end
9$orb
101*2
111
122
13$end
14$geo
15H 0.0 0.0 0.0
16H 0.0 0.0 0.74
17$end
在输出文件中,我们可以看到如下信息,提示用户这是一个态平均计算:
1 Stat average computation with folllowing states and weights:
2
3 STATE WEIGHT
4 1 0.700
5 2 0.300
最终,除了总能量以外,XMVB 还会输出态平均中每个涉及的电子态的能量,结构系数及权重信息,如下所示(以基态信息为例):
1 STATE: 1
2 ENERGY: -1.13344128
3 WEIGHT: 0.70000000
4
5 ****** COEFFICIENTS OF STRUCTURES ******
6
7 1 -0.90250405 ****** 1-2
8 2 0.05647560 ****** 1 1
9 3 0.05647560 ****** 2 2
10
11
12 ****** WEIGHTS OF STRUCTURES ******
13
14 1 0.90219477 ****** 1-2
15 2 0.04890261 ****** 1 1
16 3 0.04890261 ****** 2 2