2. 价键结构
我们将在这一讲中介绍价键结构的相关概念,了解什么是价键结构,以及如何为一个给定体系产生价键结构。
在开始今天的内容之前,请大家思考一下:
思考
H2 有几个价键结构?1,3-丁二烯呢?更复杂一些,萘呢?O2 分子呢?
2.1. 活性空间
让我们先回到分子轨道理论。分子轨道理论的基础是 Hartree-Fock( HF )方法。在 HF 方法中,分子的波函数由单个 Slater 行列式描述,电子依据能量最低原理、泡利不相容原理和洪特规则占据分子轨道。当电子从低能级轨道向高能级轨道激发,就产生了激发行列式,如 图 2.1.1 所示。
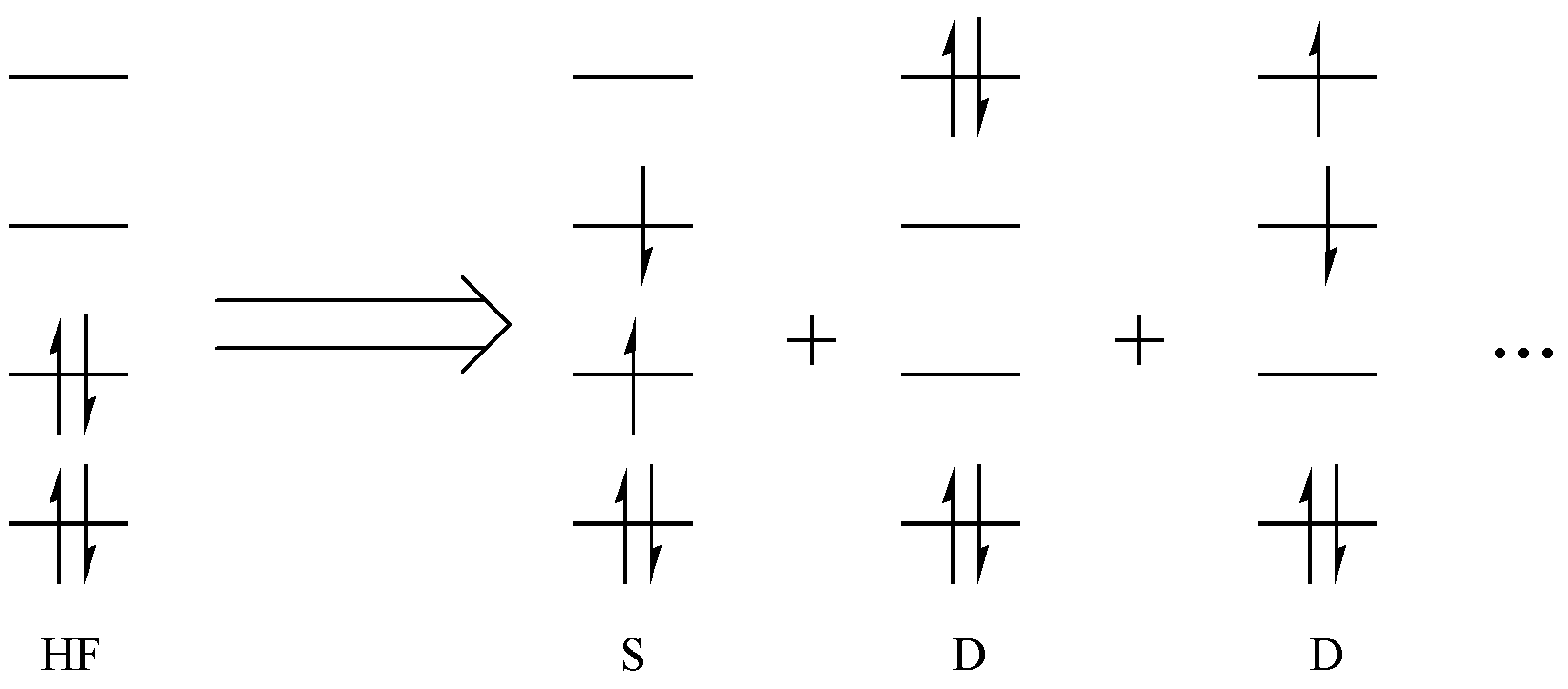
图 2.1.1 从 HF 波函数产生激发行列式
如果只有一个电子产生激发,就是所谓的单激发( S );如果有两个电子激发,就是双激发( D )。以此类推,对于一个 n 电子体系,最多会有 n 级激发。如果我们将所有的 n 级激发全都考虑进去,然后求解总波函数,就是完全组态相互作用( Full CI, FCI )方法。FCI 方法是公认的最精确的方法,只是这种方法计算量太大,只有少数简单体系可以进行 FCI 计算。于是,化学家们退而求其次,针对体系激发的特点,对于某个化学问题,只允许一部分电子在一部分轨道间进行完全激发。这一部分电子和轨道就被称为 活性空间 ,相应的电子和轨道分别被称为 活性电子 和 活性轨道 ,如 图 2.1.2 中红色部分所示。相对应的,剩余的占据轨道和电子不参与激发,在激发过程中保持不动(占据状态不变),称为 非活性空间 ,如 图 2.1.2 中的蓝色部分。
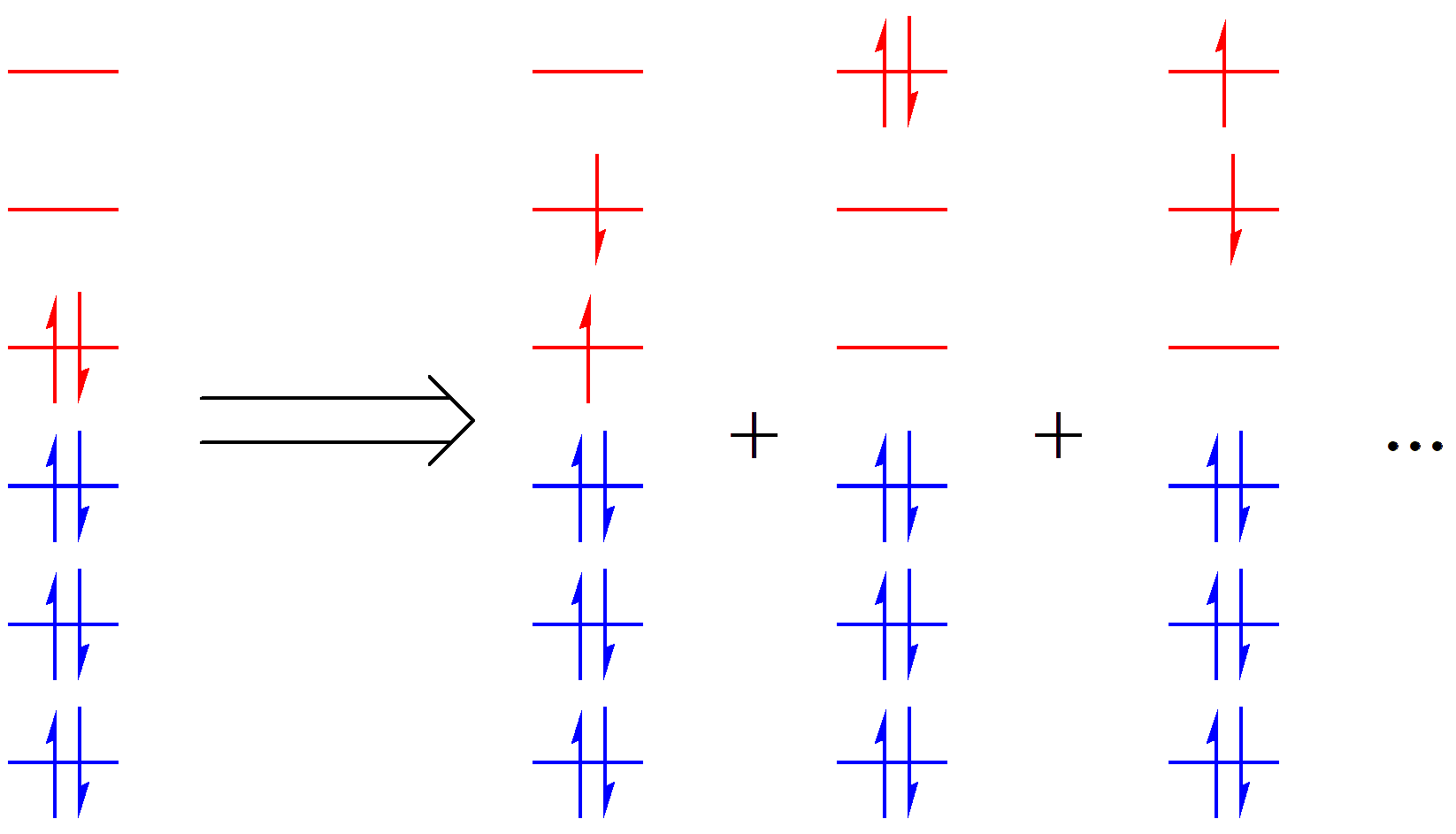
图 2.1.2 分子轨道理论中的活性空间表示
价键理论中的活性空间和分子轨道类似。在价键理论中,依据电子在价键轨道中的不同占据,我们可以得到一系列价键结构。我们针对特定的体系和化学问题,只选取与化学问题有关的价键轨道和对应的电子来产生价键结构,这部分轨道和电子就称为 活性空间 ,如 图 2.1.3 中 C2 H6 分子价键结构中的红色和蓝色部分分别表示活性和非活性空间。
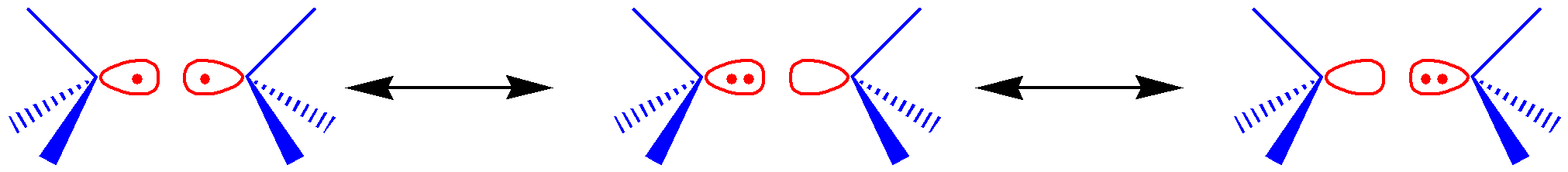
图 2.1.3 价键理论中的活性空间表示
活性空间一般用 (m , n) 来表示,其中 m 表示活性电子数, n 表示活性轨道数。比如, (2,2) 表示 2 个活性电子和 2 个活性轨道; (8,6) 表示 8 个活性电子和 6 个活性轨道。需要注意的是,非活性空间永远是闭壳层。因此,对于含有未成对电子(自旋多重度大于 1 )的体系,未成对电子和相应的轨道也需要包含在活性空间内。
活性空间是一个非常基础也非常重要的概念。不仅仅在价键理论中,分子轨道理论中的多组态自洽场( MCSCF )方法,如完全活性空间自洽场( CASSCF )和限制活性空间自洽场( RASSCF )等方法中同样也需要活性空间来指明电子激发的数量和范围。
2.2. Rumer 规则
现在,我们已经知道了活性空间的概念。那么,对于一个体系(如一个分子),当指定了活性空间,我们需要如何产生所需的结构呢?
比如,对于 H2 分子,我们可以选择 2 个活性电子和 2 个活性轨道的方案,这样我们就可以得到下图的 3 个价键结构:
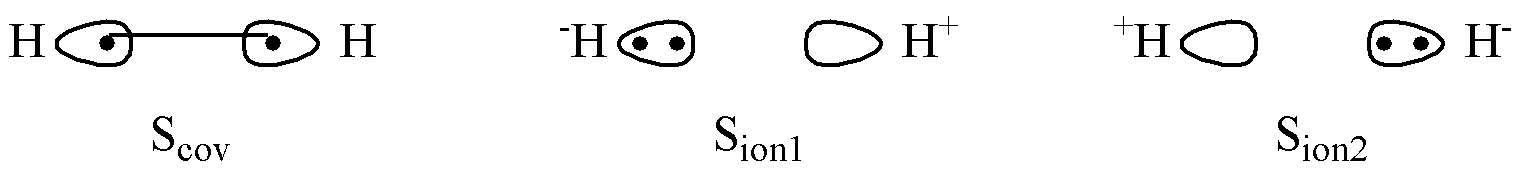
这个是非常直观且自然的。对于HF、F2 、C2 H6 等体系,只要我们选取的是两个原子或基团中的单键,我们都可以得到类似的 3 个价键结构。那么,如果是更复杂的情况呢?
对于 1,3-丁二烯( C4 H6 ),它的分子结构如下:
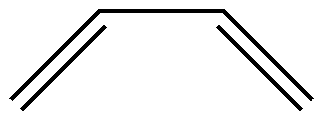
假设活性空间是 4 个 \(\pi\) 电子和对应的轨道,我们如何获得所有的价键结构? 一个最简单的方案就是将所有的可能行都列出来。根据排列组合,我们可以获得相应的 3 个共价结构:
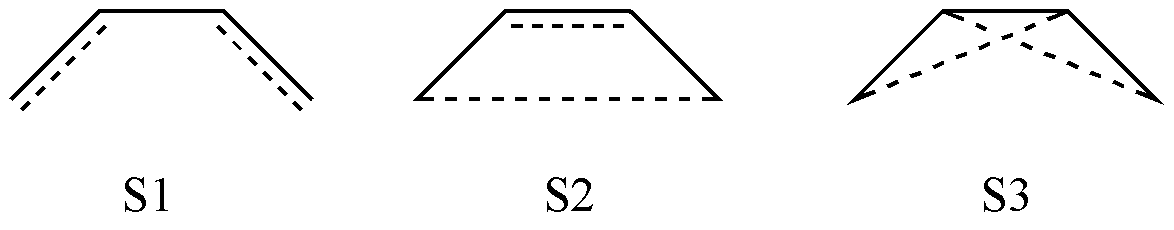
图 2.2.1 C4 H6 分子中可能的共价结构
但是,如果你利用这 3 个价键结构进行计算,会发现计算出错了!这是因为这 3 个结构并不是线性独立的。可以证明,S3 可以表示为 S1 和 S2 的线性组合:
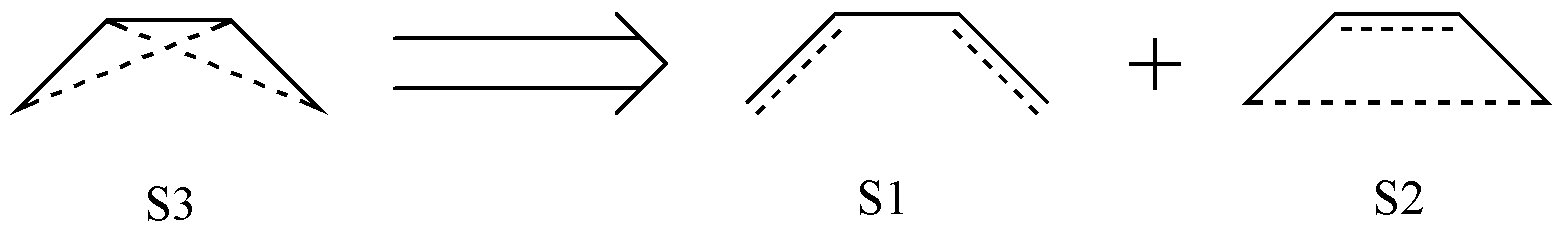
图 2.2.2 C4 H6 分子共价结构间的线性组合
实际上,C4 H6 分子一共只有 2 个共价结构。因此,我们按照简单的排列组合获得的价键结构其实是有 冗余 的,我们必须要将冗余的结构剔除才能进行价键计算。那么,我们如何识别冗余结构?或者说,我们如何产生 没有冗余 的价键结构?这就需要我们运用 Rumer 规则 来产生价键结构。
2.2.1. 闭壳层情况
我们先来看简单的闭壳层情况。对于闭壳层体系,所有电子完全配对,此时 Rumer 规则产生价键结构的流程如下:
将活性轨道按一定顺序排列为一圈,并标记序号;
对于离子结构,确定双占轨道和空轨道的位置;
对剩余的轨道,按照排列组合两两配对划线,并剔除所有 交叉 的结构,剩下的就是线性独立的结构。
让我们尝试用 Rumer 规则来产生 C4 H6 的价键结构。首先,我们将 4 个活性轨道排列为一圈并标记序号:
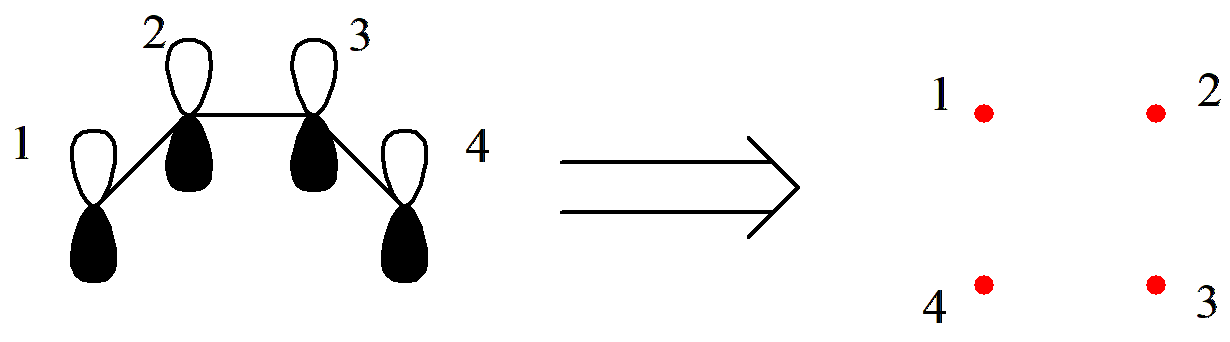
图 2.2.3 将 C4 H6 的 \(\pi\) 轨道按顺序排列
对于共价结构,我们直接将 4 个轨道两两配对划线:
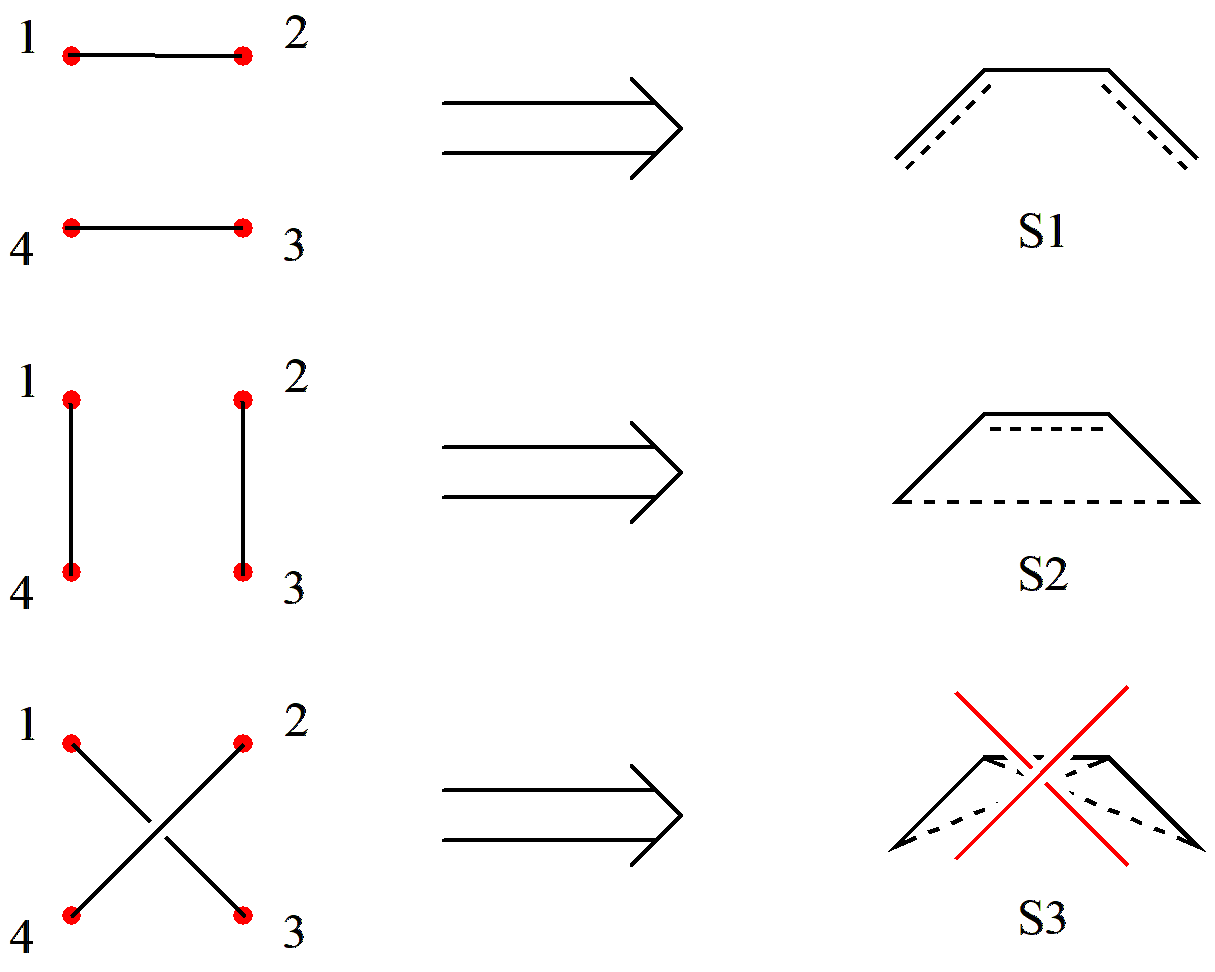
图 2.2.4 配对成键,并剔除“交叉”的情况
由于 S3 出现了连线的 交叉 ,因此需要剔除。所以,我们得到了 2 个共价结构。
对于离子结构,假设我们现在有一个双占轨道和一个空轨道(一个离子对),因此我们先要固定离子对的位置。由于离子对不是共价键,因此不受“不交叉”规则的影响,按照简单的排列组合来整理即可。最终,我们可以得到如 图 2.2.5 所示的 20 个价键结构,包括 2 个共价结构( S1-S2 ),12 个一级离子结构(含有一对离子对, S3-S14 )和 6 个二级离子结构(含有 2 对离子对, S15-S20 )。
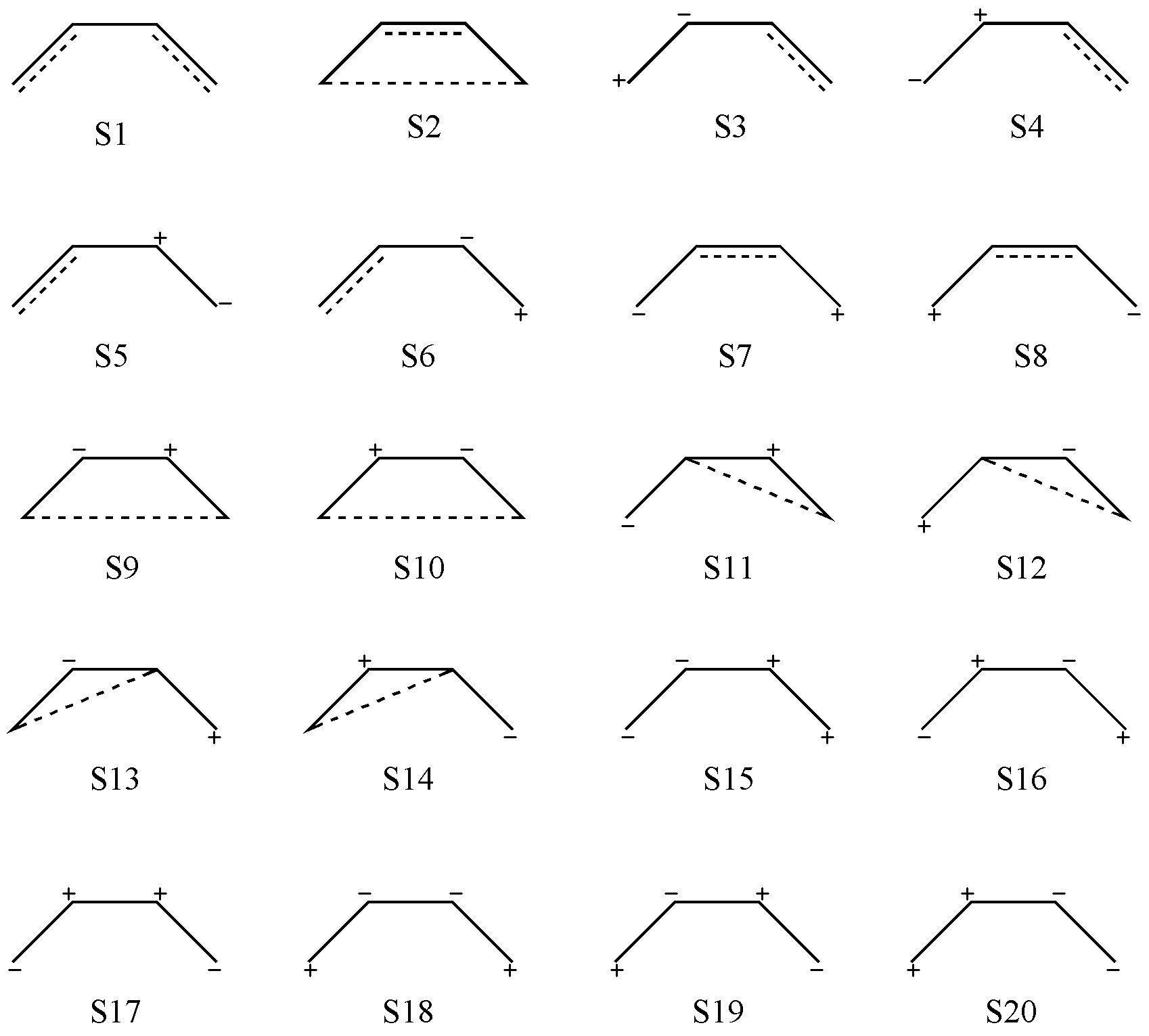
图 2.2.5 经 Rumer 规则产生的 C4 H6 全部价键结构
2.2.2. 开壳层情况
实际计算中,我们会遇到很多含有未成对电子的开壳层体系,比如各种自由基。对于这类体系,我们需要对闭壳层的 Rumer 规则做一些修改:
将活性轨道围成一圈时,我们需要多加一个 极点 。
在连线时,极点只和未成对电子连线,且一个极点可以和多个未成对电子连线;
共价键既不可以和其它共价键交叉,也不可以和涉及极点的线交叉。
我们再用 C4 H6 的共价结构举个例子。假设此时 C4 H6 是三重态,即有 2 个 \(\pi\) 电子是未成对的。因此,我们第一步画的圈就变成了:
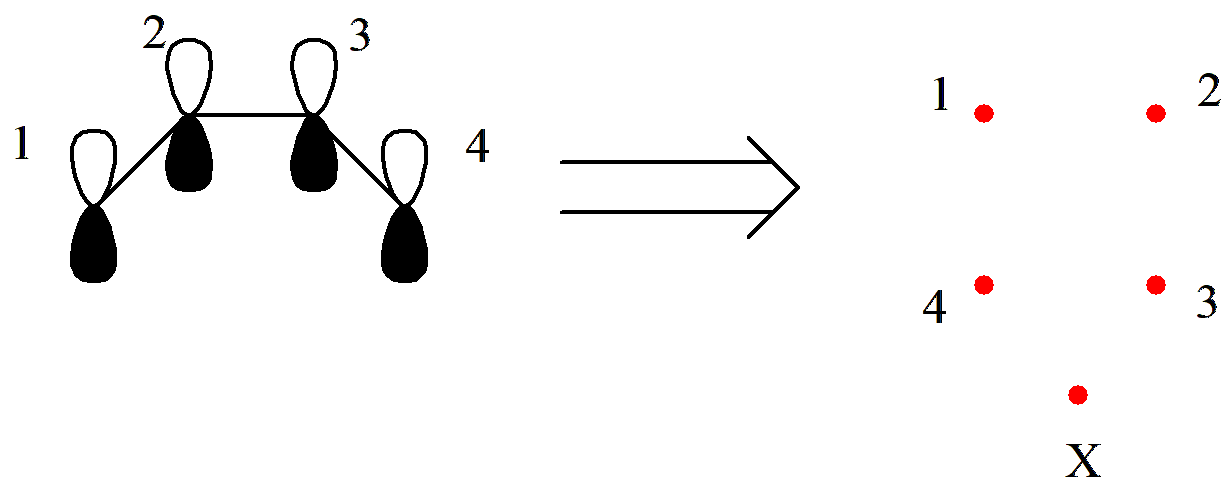
图 2.2.6 三重态 C4 H6 的 \(\pi\) 轨道排列
其中 X 表示极点,用来和未成对电子连线。之后,我们进行连线:
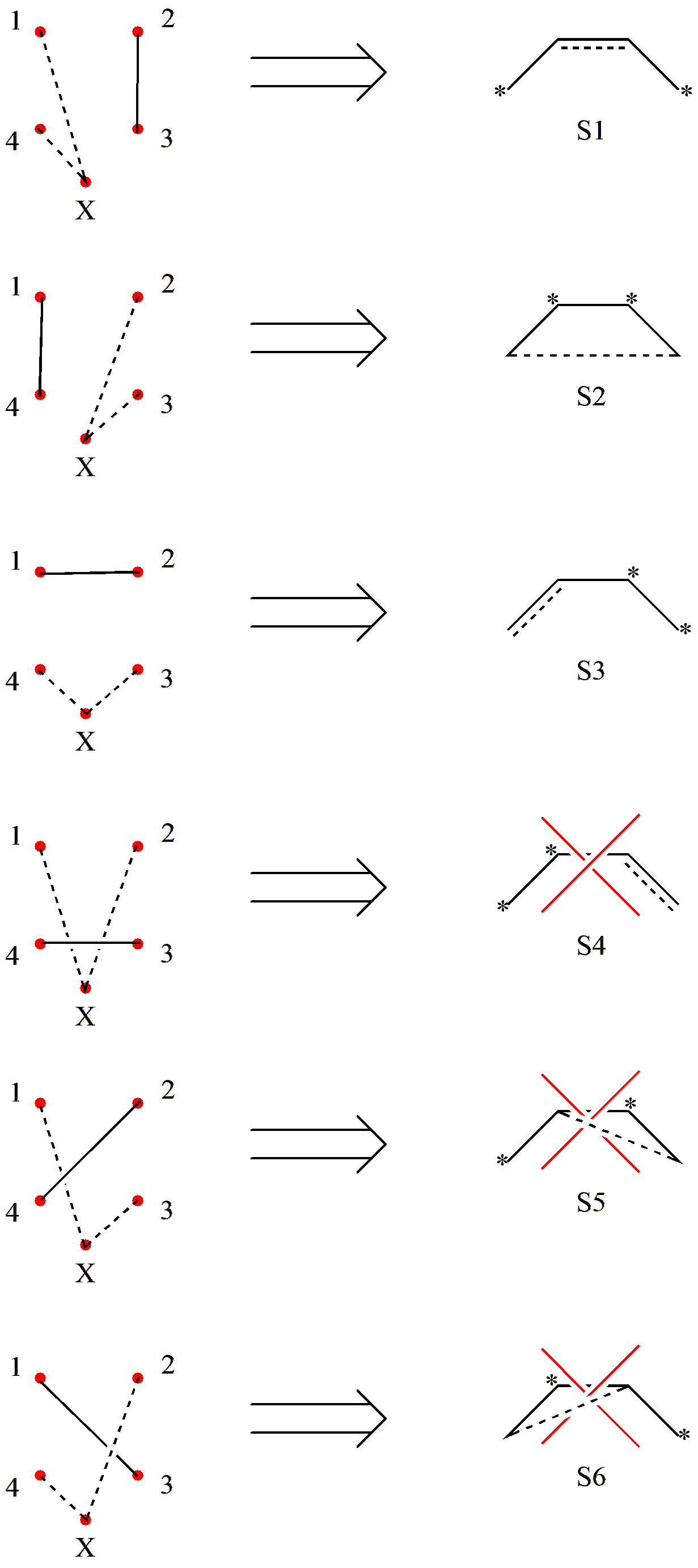
图 2.2.7 三重态 C4 H6 的共价结构筛选
可以看到,虽然我们通过排列组合得到了6个共价结构,但是只有 3 个是满足“不交叉”的要求的。因此,三重态 C4 H6 有 3 个共价结构。同样的,我们可以根据 Rumer 规则得到三重态 C4 H6 所有的价键结构。
练习
请用 Rumer 规则画出:
三重态 C4 H6 所有的价键结构。
闭壳层苯分子 \(\pi\) 空间 (6,6) 下的所有价键结构。
丙烯基自由基( C3 H5 ) \(\pi\) 空间 (3,3) 下所有的价键结构,结构见下图。

实际计算中,XMVB 可以根据 Rumer 规则自动产生指定活性空间下的价键结构。大家在这里只需要了解 Rumer 规则的思想就可以了,无需过多纠缠于细节。
2.3. 如何解读 XMVB 中的价键结构?
在 XMVB 计算中,每个价键结构会被表示为一串数字。那么,如何解读这些数字,从而知道我们得到了哪些价键结构?
在 XMVB 中,价键结构的表示以电子对为基础。从第一数字开始,每两个数字表示一对电子的占据情况,数字表示相应电子的占据轨道编号。对于含有 a 个未成对电子的体系(多重度 a+1 ),结构的最后 a 个数字表示未成对电子的占据轨道编号。需要说明的是,价键结构的解读需要结合轨道的定义才能真正完成,因为结构的化学图像依赖于具体的轨道。我们这里先解释价键结构本身的解读,价键轨道的定义以及如何进一步完成解读将在后续章节完成。
我们首先以 H2 为例。H2 是闭壳层体系,所有电子都成对。XMVB 的输出显示,H2 包含以下 3 个价键结构:
1 Number of structures: 3
2
3 The following structures are used in calculation:
4
5 1 ***** 1-2
6 2 ***** 1 1
7 3 ***** 2 2
第一个结构 1-2 表示一对电子,其中一个占据在轨道 1 上,另一个电子占据在轨道 2 上。中间的 - 表示这两个电子配对成键,因此这个结构表示H2的共价结构。结构 1 1 表示电子对的两个电子都占据在同样的轨道 1 上,因此表示一个离子结构。同理,结构 2 2 表示另一个离子结构。
我们再来看一个复杂点的体系,F2 分子。F2 分子也是闭壳层,总共有 18 个电子,活性空间为 (2,2)。简单计算可知,非活性轨道一共有 8 个,非活性电子有 16 个。XMVB 给出的结构信息为:
1 Number of Structures: 3
2
3The following structures are used in calculation:
4
5 1 ***** 1:8 9-10
6 2 ***** 1:8 9 9
7 3 ***** 1:8 10 10
可以看到,每个结构开头都有 1:8 这个信息。在 XMVB 中,这个信息表示有 8 对电子双占,这些电子占据了前 8 个轨道。这种写法等同于 1 1 2 2 3 3 4 4 5 5 6 6 7 7 8 8 。由于每个结构都有这个信息,说明这 16 个电子在所有结构中的占据都是一样的,因此表示的是非活性空间。第一个结构最后的 9-10 表示两个电子分别占据轨道 9 和 10 且配对成键,是共价结构。后两个结构分别表示两个离子结构。
最后,我们来看看 O2 分子的结构解读。由于 O2 的结构比较多,因此我们选取了以下两个结构:
1 1 ***** 1:4 7 7 10 10 5-6 8 9
2 2 ***** 1:4 8 8 9 9 5-6 7 10
O2 分子是三重态,因此每个结构最后两个数字表示未成对电子占据的轨道。每个结构开头都有 1:4 ,表示前 4 对电子在所有结构中都是双占,因此是非活性空间。第一个结构中的 7 7 10 10 表示两对电子分别双占轨道 7 和 10 , 5-6 表示一对电子分别占据轨道 5 和 6 并成键,最后轨道 8 和 9 分别占据一个未成对电子。同样,我们可以发现,第二个结构同样表示轨道 5 和 6 之间成键,但是双占轨道变成了 8 和 9 ,未成对电子占据了轨道 7 和 10 。
2.4. 哪些价键结构比较重要?
在价键计算中,我们常常需要判断哪些价键结构比较重要。例如,当我们需要判断化学键属于哪种类型(共价、离子或其它),我们需要了解其中最重要的价键结构。此外,当计算涉及的价键结构非常多的时候,我们常常需要对价键结构进行筛选,从这些价键结构中挑选出一部分价键结构进行计算,而根据价键结构的重要性进行挑选就是最常用的筛选方案。在价键计算中,我们常常使用 权重 作为依据判断结构的重要性。
价键结构 K 的系数 \(C_K\) 可以通过求解久期方程来得到:
其中 H 和 M 分别为 Hamiltonian 和重叠矩阵
其中 \(\Phi_K\) 和 \(\Phi_L\) 表示价键结构 K 和 L 。
在分子轨道理论中,由于轨道正交,结构(组态)之间一定是正交的,因此,权重可以简单地表示为系数的平方:
对于价键理论,由于轨道不正交,我们不能简单地用上式来表示权重。此时,权重的定义并不唯一。常用的权重有 Coulson-Chirgwin(CC)权重、Löwdin 权重、Inverse 权重等。
CC 权重是最常用的权重类型,定义直观,计算简便。它的定义如下:
CC 权重的问题在于某些情况下会出现权重为负数的情况。一般来说,负权重意味着这个结构不重要。Löwdin 权重的定义为:
Inverse 权重的定义为:
其中 \(N_K\) 的定义为:
这两种权重定义都避免了负权重的产生,但是都需要对重叠矩阵进行求逆或对角化操作,因此计算量比 CC 权重更大。XMVB 中同时给出了以上这几种权重定义的结果,用户可以根据自己的实际需要选取合适的权重。