6. Excited States
Up to this point, our focus has been on ground-state calculations. In real research tasks, however, molecules cannot always remain in the ground state, and researchers’ interests are not confined to ground-state properties alone. Therefore, in this section we will briefly discuss how to carry out calculations of excited states.
6.1. Different Pictures of Excited States in MO and VB Theories
In MO theory, atomic orbitals are linearly combined to form molecular orbitals. Each molecular orbital has its own energy, and electrons occupy orbitals according to the aufbau principle, i.e., filling them from lowest to highest energy. In the case of orbital degeneracy, Hund’s rule applies: electrons are first distributed with parallel spins among the degenerate orbitals before pairing up. The electronic state obtained in this manner is typically the ground state, since all unoccupied orbitals are of higher energy than the occupied ones. If, however, electron occupation does not follow these rules—for example, if in degenerate orbitals one orbital is filled completely before occupying the others, or if a higher-energy orbital is filled while a lower-energy orbital remains empty or only partially filled—the total energy of the molecule increases. The resulting higher-energy state is what we call an excited state. Such states are unstable and are usually formed when electrons in a ground-state molecule absorb some form of energy (e.g., photons), which promotes them into higher-energy orbitals or configurations. In MO theory, the excitation energy of a molecule can be roughly estimated by Koopmans’ theorem, namely
In actual calculations, however, because orbital occupation changes after excitation, the orbitals themselves undergo relaxation. Therefore, the true excitation energy must take orbital relaxation into account, and a self-consistent field iteration is required.
In VB theory, the molecular wavefunction is determined by two aspects: the choice of orbitals that define the resonance structures, and the linear combination of these structures. Different orbital choices correspond to different physical meanings and chemical pictures of the resonance structures, while different combination coefficients yield different properties and states of the molecule. The combination coefficients are obtained by solving the secular equation. For a molecule with n structures, the secular equation corresponds to an n-dimensional generalized eigenvalue problem, which yields n solutions in total. The first solution corresponds to the lowest-energy combination. When orbital occupations follow the aufbau principle, this first root corresponds to the ground state of the molecule. If we do not take the first root, or if the electron occupations do not follow the lowest-energy filling, the resulting combination of structures changes, and we obtain an excited state. We can see here that excited-state calculations in VB theory involve two dimensions: the choice of orbitals and the variation in combination coefficients. Since orbital choices determine the specific physical meaning and chemical picture of the resonance structures, please refer back to Section 2 and Section 3. In this lecture, our focus will be on how changes in combination coefficients affect excited states.
6.2. How to Calculate Excited States in XMVB
6.2.1. Obtaining Specific Excited States
XMVB provides the keyword NSTATE=IROOT to select different roots of the secular equation, where IROOT is a non-negative integer indicating which excited state to compute. NSTATE=1 means the program will select the second root to obtain the first excited state, NSTATE=2 corresponds to the second excited state, and so forth. NSTATE=0 specifies the ground-state (zeroth excited state) calculation, which is also the default.
Let us first look at the H2 molecule to see the effect of NSTATE=IROOT. By default, XMVB performs a ground-state calculation. For an H–H bond length of 0.7 Å with the cc-pVDZ basis set in an HAO calculation, the energy obtained is –1.14386 a.u., and the coefficients and weights of the three valence-bond structures are as follows:
1 ****** COEFFICIENTS OF STRUCTURES ******
2
3 1 -0.84426638 ****** 1-2
4 2 -0.09228443 ****** 1 1
5 3 -0.09228443 ****** 2 2
6
7
8 ****** WEIGHTS OF STRUCTURES ******
9
10 1 0.84327776 ****** 1-2
11 2 0.07836112 ****** 1 1
12 3 0.07836112 ****** 2 2
Here, the covalent structure clearly dominates. If we add NSTATE=1, the final energy becomes –0.38420 a.u., and the corresponding structure coefficients and weights are:
1 ****** COEFFICIENTS OF STRUCTURES ******
2
3 1 -0.00000000 ****** 1-2
4 2 1.51521437 ****** 1 1
5 3 -1.51521436 ****** 2 2
6
7
8 ****** WEIGHTS OF STRUCTURES ******
9
10 1 0.00000000 ****** 1-2
11 2 0.50000000 ****** 1 1
12 3 0.50000000 ****** 2 2
We can see that the covalent structure has vanished, and the coefficients of the ionic structures have changed from equal to opposite. This corresponds to the coefficients of the first excited state.
Next, let us consider a more complex example: ₂O2. The triplet state of O2 requires 12 resonance structures, while the singlet state requires 10. The 12 structures of the triplet state are shown in Fig. 6.2.1, with T1 and T2 being the most important.
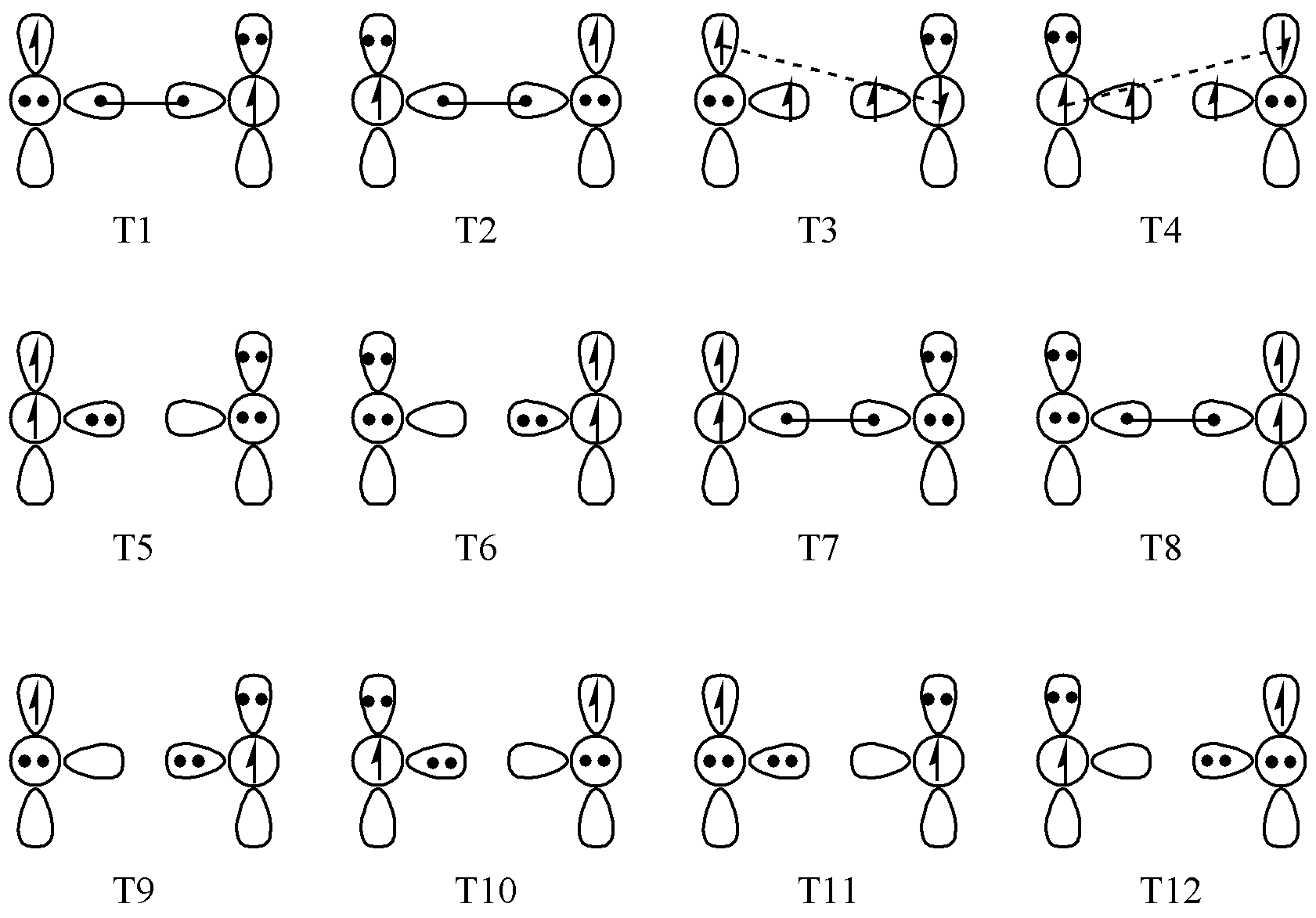
Fig. 6.2.1 The 12 valence-bond structures of triplet O2
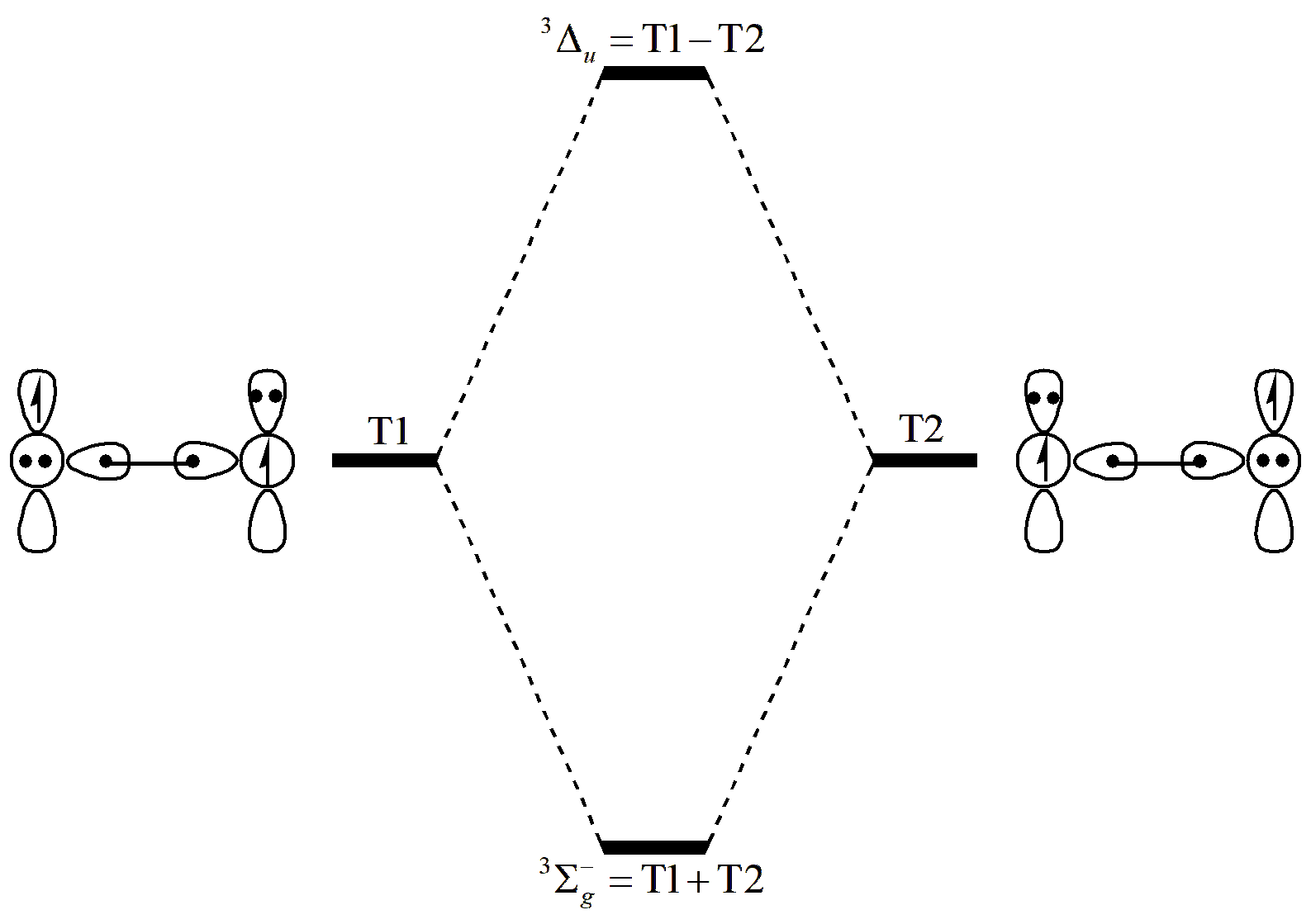
Fig. 6.2.2 The ground and excited states of triplet O2
As shown in Fig. 6.2.2, different combinations of T1 and T2 correspond to different electronic states. In the ground state, the coefficients of these two structures are the same; in the excited state, the coefficients are opposite.
To obtain the excited state of the triplet, we can compute the first excited state by setting NSTATE=1, as shown below:
1O2 L-VBSCF
2$ctrl
3vbscf nstr=12 nao=6 nae=8 iscf=5 iprint=3
4orbtyp=hao frgtyp=sao nmul=3 itmax=200 nstate=1
5int=libcint basis=cc-pvdz
6guess=read
7$end
8$str
91:4 7 7 10 10 5 6 8 9
101:4 8 8 9 9 5 6 7 10
111:4 7 7 10 10 8 9 5 6
121:4 8 8 9 9 7 10 5 6
131:4 7 7 9 9 5 6 8 10
141:4 8 8 10 10 5 6 7 9
151:4 8 8 10 10 5 5 7 9
161:4 7 7 9 9 6 6 8 10
171:4 8 8 9 9 5 5 7 10
181:4 7 7 10 10 6 6 8 9
191:4 7 7 10 10 5 5 8 9
201:4 8 8 9 9 6 6 7 10
21$end
22$frag
231*6
24spzdxxdyydzzfzzzfxxzfyyz 1
25spzdxxdyydzzfzzzfxxzfyyz 2
26pxdxzfxxxfxyyfxzz 1
27pxdxzfxxxfxyyfxzz 2
28pydyzfyyyfxxyfyzz 1
29pydyzfyyyfxxyfyzz 2
30$end
31$orb
321*10
331
342
351
362
371
382
393
404
415
426
43$end
44$geo
45O 0.0 0.0 0.0
46O 0.0 0.0 1.2
47$end
48$gus
49 8 8 8 8 8 8 3 3 3 3
50 1.0019774038 1 0.0065168066 2 0.0001788772 3 0.0010309980 6
51-0.0003633198 9 -0.0027414664 10 -0.0030686413 13 -0.0030026561 15
52 1.0021643892 16 0.0072505362 17 0.0002382456 18 -0.0011188449 21
53 0.0003834680 24 -0.0030102480 25 -0.0031503232 28 -0.0032525782 30
54 0.0982223750 1 -0.2504910231 2 -0.8206750714 3 -0.0401288970 6
55-0.3250457745 9 0.0663093980 10 0.0645475585 13 -0.0255293114 15
56 0.1023941578 16 -0.2356452123 17 -0.8258612653 18 0.0419646005 21
57 0.3491578062 24 0.0683659578 25 0.0684486346 28 -0.0278018600 30
58-0.0534466425 1 -0.2957625003 2 -0.0041914149 3 0.2677956458 6
59 0.7578794938 9 -0.0782023209 10 -0.0786400588 13 0.0255903120 15
60-0.0521455763 16 -0.3057336084 17 -0.0199773005 18 -0.2554552086 21
61-0.7557006120 24 -0.0788914259 25 -0.0791889215 28 0.0231651215 30
62 0.5315910156 4 0.6213802169 7 0.0117253831 12
63 0.5179245322 19 0.6342323659 22 -0.0135520711 27
64 0.5280659286 5 0.6247144659 8 0.0122018946 14
65 0.5244186959 20 0.6281355341 23 -0.0136783062 29
66$end
Compared with the ground-state calculation, the only change in the input file is the inclusion of the NSTATE keyword. Removing NSTATE=1 reverts the calculation to the ground state. The final results are given in Table 6.2.1:
Structure |
Ground State |
Excited State |
||
|---|---|---|---|---|
Coefficient |
Weight |
Coefficient |
Weight |
|
T1 |
-0.343 |
0.220 |
-0.492 |
0.327 |
T2 |
-0.343 |
0.220 |
0.492 |
0.327 |
T3 |
-0.018 |
0.000 |
-0.036 |
0.001 |
T4 |
0.018 |
0.000 |
-0.036 |
0.001 |
T5 |
0.166 |
0.073 |
0.000 |
0.000 |
T6 |
0.166 |
0.073 |
0.000 |
0.000 |
T7 |
0.178 |
0.091 |
0.000 |
0.000 |
T8 |
0.178 |
0.091 |
0.000 |
0.000 |
T9 |
-0.125 |
0.058 |
-0.180 |
0.086 |
T10 |
-0.125 |
0.058 |
0.180 |
0.086 |
T11 |
-0.125 |
0.058 |
-0.180 |
0.086 |
T12 |
-0.125 |
0.058 |
0.180 |
0.086 |
From these results, we can see that the combination of coefficients of T1 and T2 indeed changes between the ground and excited states. We also see that, both in terms of coefficients and weights, the excited state is dominated by T1 and T2, with diminished contributions from the other structures. The first excited state of triplet O2 is thus primarily composed of the two structures T1 and T2.
6.2.2. State-Averaged Calculations
When performing excited-state calculations, it is often useful to employ state averaging. In a state-averaged calculation, we do not optimize a single electronic state. Instead, we combine the energies of several states of interest according to predefined weights, and then use self-consistent field iterations to optimize the orbitals such that the weighted total energy is minimized:
where \(E\) is the total energy, \(E_i\) is the energy of a particular state included in the state averaging, and \(w_i\) is the corresponding weight, which remains fixed during the optimization.
In XMVB, the keyword WSTATE is used to perform state-averaged calculations. Its syntax is:
WSTAE( start )= \(w_{\textrm{start}}\), \(w_\textrm{start+1}\), …
Here, start in parentheses denotes the index of the first state included. The ground state is indexed as 1, the first excited state as 2, and so forth. The numbers after the equals sign, separated by commas, specify the weights of consecutive states. For example:
WSTATE(1)=0.4,0.6
performs a two-state average, where the ground state has weight 0.4 and the first excited state has weight 0.6. Multiple WSTATE keywords can also be combined, for example:
WSTATE(1)=0.5,0.5 WSTATE(5)=0.4,0.6
Here, the state averaging involves four states: the ground state (weight 0.5), the first excited state (weight 0.5), the fourth excited state (weight 0.4), and the fifth excited state (weight 0.6). Note that the sum of weights here is greater than 1. XMVB automatically normalizes the weights to unity, so the above input is equivalent to:
WSTATE(1)=0.25,0.25 WSTATE(5)=0.2,0.3
Users only need to focus on the ratios between the weights of different states. The default value of WSTATE is
WSTATE(1)=1.0,0.0,0.0, ...
Which means only the ground state is optimized. State-averaged calculations can be applied in both VBSCF and BOVB computations. As an example, consider H2. We perform a state-averaged calculation involving the ground state and the first excited state, with weights of 0.7 and 0.3, respectively. The input file is:
1H2 State average
2$ctrl
3vbscf
4str=full nao=2 nae=2 iscf=5
5orbtyp=hao frgtyp=atom
6int=libcint basis=cc-pvdz
7wstate(1)=0.7,0.3
8$end
9$orb
101*2
111
122
13$end
14$geo
15H 0.0 0.0 0.0
16H 0.0 0.0 0.74
17$end
In the output file, we see a message confirming that this is a state-averaged calculation:
1 Stat average computation with folllowing states and weights:
2
3 STATE WEIGHT
4 1 0.700
5 2 0.300
Finally, in addition to the total energy, XMVB also outputs the energy, structure coefficients, and weights for each individual state included in the averaging. For example, the ground-state information is:
1 STATE: 1
2 ENERGY: -1.13344128
3 WEIGHT: 0.70000000
4
5 ****** COEFFICIENTS OF STRUCTURES ******
6
7 1 -0.90250405 ****** 1-2
8 2 0.05647560 ****** 1 1
9 3 0.05647560 ****** 2 2
10
11
12 ****** WEIGHTS OF STRUCTURES ******
13
14 1 0.90219477 ****** 1-2
15 2 0.04890261 ****** 1 1
16 3 0.04890261 ****** 2 2